PATOGÉNESIS Y ETIOLOGÍA
Lo que los científicos sí saben es que el sistema inmunológico del cuerpo comienza a atacar al propio cuerpo, lo que se conoce como una enfermedad autoinmunológica. En el Síndrome de Guillain-Barré, no obstante, el sistema inmunológico comienza a destruir la cobertura de mielina que rodea a los axones de muchos nervios periféricos, o incluso a los propios axones (los axones son extensiones delgadas y largas de las células nerviosas que transmiten las señales nerviosas.
En enfermedades en las que los recubrimientos de mielina de los nervios periféricos son lesionados o quedan afectados, los nervios no pueden transmitir señales con eficiencia. A ello se debe el que los músculos comiencen a perder su capacidad de responder a los mandatos del cerebro, mandatos que han de transportarse a través de la red nerviosa.
Las evidencias en la mayoría de los estudios sobre síndrome de Guillain-Barré (SGB) sugieren que las manifestaciones clínicas de esta afección son el resultado de una afección inmunológica medida por células dirigidas hacia el nervio periférico.
En el síndrome de Guillain-Barré (SGB) grave, los axones se dañan tan gravemente que se cortan en el punto en que se inflaman. La parte distal (la parte del lado alejado del centro) vuelve a crecer desde el centro hacia el exterior. La tasa de crecimiento se estima que es de 1 mm. al día. Puesto que los nervios más largos del cuerpo humano tienen aproximadamente un metro de longitud, podría esperarse que se requirieran casi tres años para que se regeneren los nervios más largos si se dividen cerca de sus cuerpos celulares. En la práctica, el nuevo crecimiento y la recuperación son con frecuencia incompletos
Los investigadores han identificado como agentes potenciales que propagan el síndrome de Guillain-Barré (SGB) los siguientes puntos:
Campylobacter jejuni
Virus de Varicela-zoster
Micoplasma Pneumoniae
Epstein Barr
Herpes Virus
El riesgo para desarrollar el síndrome de Guillain-Barré (SGB) después de recibir una inmunización es bajo; los casos raros de SBG han ocurrido después de la vacunación contra la rabia o H1N1.
El síndrome de Guillain-Barré puede presentarse en asociación con infecciones virales como mononucleosis, SIDA y herpes simple o después de infecciones con bacterias como micoplasma y algunos tipos de diarrea.
No todos los desencadenantes son infecciosos. Existen otros eventos como la cirugía y los traumatismos, los cuales sólo se encuentran asociados en un pequeño porcentaje (2-3%).
También algunas enfermedades malignas como la leucemia no linfoblástica y la leucemia linfocítica crónica se han asociado con el desarrollo del síndrome de Guillain-Barré (SGB); probablemente la autoinmunidad también está involucrada.
Es necesario considerar que el cuadro infeccioso puede ser inespecífico o incluso subclínico.
Waksman y Adams demostraron que una enfermedad del nervio periférico clínica y patológicamente indistinguible del síndrome de Guillain-Barré (SGB) se puede desarrollar en animales alrededor de las dos semanas después de la inmunización del nervio periférico (neuritis alérgica experimental (NAE). Brostoff y colegas sugirieron que el antígeno en esta reacción es la proteína básica de la mielina, designada como proteína P2, encontrada solamente en la mielina del nervio periférico. Otras investigaciones subconsecuentes indicaron que un factor neuritigénico era un péptido específico en la proteína P2. Posteriormente ha quedado claro que no existe una reacción antígeno-anticuerpo unificada y que un número de elementos de la mielina y quizás axonales pueden estar involucrados a la reacción inmune. Los cambios patológicos que se suceden en este proceso se desarrollan en 4 etapas o momentos según Asbury y colaboradores.
- Linfocitos que se adhieren a la pared de los vasos endoneurales y migran a través de pared vascular engrasándola y transformándola.
- Más linfocitos han migrado dentro de los tejidos vecinos. El primer efecto sobre los nervios de la ruptura de la mielina mientras el axón está respetado (desmielinización segmentaria)
- La lesión es más intensa. Presencia de leucocitos polimorfonucleares y de linfocitos. Hay una interrupción del axón en adición al daño de la mielina, como resultado, los músculos desarrollan atrofia por denervación y el cuerpo de la célula nerviosa muestra cromatolisis central. Si el daño axonal es distal la célula nerviosa sobrevivirá y se alcanza la regeneración y la recuperación clínica probablemente.
- Interrupción axonal. Ha ocurrido próximamente porque ha habido una lesión nerviosa proximal o de las raíces, el cuerpo neuronal puede morir y se desarrolla disolución en esta situación no hay regeneración, solo existe la posibilidad de reinervación colateral de los músculos por las fibras motoras sobrevivientes.
Hantunny y colegas han hallado niveles elevados de interleucina (IL-2) unida a receptores provenientes de las células T activadas, y IL-2 por sí sola en el suero de pacientes con síndrome de Guillain-Barré (SGB) agudo, lo que refleja la activación de estas células, complemento que es también visto y necesario en los ataques iniciales de mielina. Aunque la trasmisión de la NAE por células T que sensibilizan a la mielina es fuerte en la evidencia de su rol en el síndrome de Guillain-Barré (SGB), los anticuerpos anti-mielina pueden jugar también una parte significativa.
El suero de los pacientes con síndrome de Guillain-Barré (SGB) daña la mielina en cultivos y puede provocar la destrucción vesicular de la misma. La inyección subespineural de suero de pacientes con síndrome de Guillain-Barré (SGB) en el nervio ciático de las ratas provoca desmielinización local y bloqueo de la conducción. Los estudios de Koski y asociados sobre el daño mielínico dependiente de complemento por anticuerpo antimielina es una IgM y proporciona la mejor evidencia de que los anticuerpos antimielina están aptos para iniciar la destrucción de esta, aún si las células T y los macrófagos ejecutan el daño finalmente. Un número de anticuerpos pueden detectarse inconscientemente en pacientes con síndrome de Guillain-Barré (SGB), pero el más prominente es anti-GM y su derivado anti-GQ1b, los que se encuentran en casi todos los pacientes con variante de Fisher.
Del 15% aproximadamente, de los pacientes que tienen anticuerpos anti-GM, en su curso temprano, los títulos más elevados se dan en los casos relacionados con Campylobacter. Resultaría entonces muy simplista pensar que el síndrome de Guillain-Barré (SGB) es una enfermedad exclusivamente inmune mediadas por células o humoral. Varios modelos animales de la enfermedad se asemejan al síndrome de Guillain-Barré (SGB) superficialmente, pero no muestran sus principales hallazgos clínicos ni patológicos.
La cuestión no contestada es que inicia la reacción antigénica en humanos. Todos los intentos de aislar el virus por microscopia electrónica han fallado y es probable que una variedad de agentes (virales, bacterianos, particularmente C. Yeyuni), ciertas vacunas y hasta un daño neural, sean todos por sí solos capaces de precipitar una respuesta inmune contra la mielina periférica autóloga en individuos susceptibles. La ocurrencia de una polineuritis en pacientes con SIDA o con infecciones precedentes a virus Epstein-Barr, o por citomegalovirus simplemente indican que ellas también pueden inducir tal respuesta, más que una etiología infecciosa viral implicada.
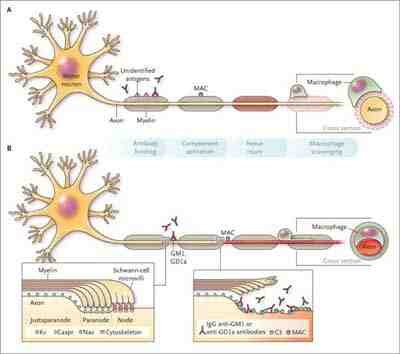
Figura 1. Inmunopatogenia del Síndrome de Guillain Barre. La invasión de macrófagos y activación del complemento conllevan al daño en la mielina.
DIAGNÓSTICO DIFERENCIAL
El síndrome de Guillain-Barré (SGB) no es solo una de las formas más frecuentes de polineuropatía vistas en los hospitales generales, sino es también la que más rápida progresa y la más potencialmente fatal. Cualquier polineuropatía que lleve al enfermo hacia la muerte por fallo respiratorio dentro de unos pocos días, es usualmente de esta variedad. No obstante, otros tipos de polineuropatías agudas que presentaremos pueden presentarse ocasionalmente y necesitan ser diferenciadas.
La situación más crítica es distinguir el síndrome de Guillain-Barré (SGB) de las enfermedades agudas de la médula espinal (marcadas por una parálisis sensorimotora por debajo de un dado nivel y afectación esfinteriana). Como una mielopatía demuestra parestesias en los dedos, puede ascender centrípetamente, lo que no es probable el síndrome de Guillain-Barré (SGB).
Otra confusión adicional puede ocurrir con las lesiones transversales agudas en las cuales los reflejos osteotendinosos están inicialmente abolidos (shock espinal), o con las mielopatías necrotizantes donde una pérdida permanente de los reflejos osteotendinosos sigue a la destrucción de las neuronas espinales. Varias reglas son útiles para distinguir el síndrome de Guillain-Barré (SGB) de la mielopatía cervical: en el síndrome de Guillain-Barré (SGB) los músculos respiratorios están usualmente involucrados, si hay parálisis generalizada, las puntas de los dedos deben estar parestésicas si los síntomas sensitivos han ascendido hasta el nivel de las mitades de las pantorrillas; es inusual la pérdida marcada de la sensibilidad proximal hasta los brazos o piernas en el estudio temprano de la enfermedad y los reflejos están invariablemente ausentes, dentro de las 24 horas, en las extremidades que están también débiles para resistir la gravedad. El hallazgo de una capacidad vital normal, colocándose sentado, en los grados de cuadriparesia se ve en las enfermedades de la médula.
Nosotros hemos observado unos pocos ejemplos de síndrome de Guillain-Barré (SGB) generalizado o de variante Fisher que han sido confundidos con trombosis de la arteria basilar. La presencia de pupilas reactivas arreflexia y anormalidades de la onda F en el primero y de reflejos vivos y signos de Babinski en el último; debe distinguir las dos afecciones. La característica más importante del síndrome de Guillain-Barré (SGB) es una parálisis predominantemente motora, por esta razón el diagnóstico diferencial debe incluir la poliomielitis, esta última se distingue por los síntomas meníngeos, fiebre, pleocitosis temprana en el líquido cefalorraquídeo (LCR) y parálisis motora arrefléxica asimétrica generalmente.
Ptosis puerperal y debilidad oculomotora son hallazgos del síndrome de Guillain-Barré (SGB) en algunos casos que pueden provocar confusión con la miastenia gravis aguda; pero en esta no hay síntomas sensitivos y se mantienen los reflejos osteotendinosos. En el síndrome de Guillain-Barré (SGB) los músculos mandibulares permanecen relativamente fuertes, mientras que en la miastenia gravis estos se afectan y producen caída de la mandíbula con apertura bucal.
El botulismo puede simular el síndrome de Guillain-Barré (SGB), pero en el primero los reflejos pupilares están ausentes tempranamente (pueden ser vistos en los casos de síndrome de Guillain-Barré (SGB) avanzados) y hay bradicardias que generalmente no se aprecian en el síndrome de Guillain-Barré (SGB). Ricketsias (tickparálisis) o afecciones transmitidas por garrapatas es una rara entidad casi imposible de distinguir en el síndrome de Guillain-Barré (SGB), a menos que exista el antecedente de contacto con garrapatas; ambas enfermedades causan una parálisis ascendente aguda, pero la pérdida sensorial no es el hallazgo principal de la ricketsiasis o parálisis por garrapata y el líquido cefalorraquídeo (LCR) R permanece normal. La ciguatera causa parestesia y debilidad aguda facio-braquial, taquipnea, iridoplejia, que dura unos pocos días.
Los trastornos musculares de todos los pacientes atendidos en la UTI necesitan siempre ser distinguidos del síndrome de Guillain-Barré (SGB). Estos incluyen la llamada polineuropatía del paciente crítico, la neuropatía acelerada del fallo renal visto en pacientes diabéticos en diálisis peritoneal, hipofosfatemia aguda de la hiperalimentación, la miopatía esteroidea, particularmente cuando se combina con parálisis neuromuscular prolongada y el efecto prolongado de las drogas bloqueadoras neuromusculares usualmente como resultado de la acumulación de metabolitos en el fallo renal y con acidosis.
TRATAMIENTO
Los puntos esenciales en la terapia de los casos severos agudos son la asistencia respiratoria y una cuidadosa atención de Enfermería hasta que la enfermedad remita naturalmente y la recuperación sea completa o así completa en la mayoría de los casos. Menos del 30% de nuestros pacientes han necesitado ventilación mecánica, pero si la condición del paciente se puede empeorar impredecible y rápidamente, el deberá ser ingresado en un centro hospitalario tan pronto como sea posible y pueda evaluarse frecuentemente su función respiratoria.
LOS CRITERIOS ESTABLECIDOS PARA INICIAR LA ASISTENCIA VENTILATORIA MECÁNICA SON LOS SIGUIENTES:
_ Capacidad vital menor de 15 mL/Kg.
_ Presión inspiratoria máxima menor de –20 cm. de agua.
_ Presión espiratoria máxima menor de 40 cm de agua.
_ Parálisis bulbar o disfagia con peligro de broncoaspiración.
_ Hipoxemia y/o hipercapnia.
La medición de la fuerza inspiratoria máxima y la capacidad vital respiratoria funciona como una guía a la cabeza del enfermo e indica la significancia de la fuerza diafragmática y la amenaza de un fallo mecánico respiratorio. Si hay una tendencia decreciente constatada, cuando estos test son repetidos en intervalos de 3-4 horas, la intubación endotraqueal debe considerarse cuando la capacidad vital alcanza entre 10-12 ml/Kg. Este grado de afectación ventilatoria ocurre antes del primer síntoma de disnea o elevación de la concentración de CO2, pero generalmente coincide con ligeras disminuciones en las concentraciones de oxígeno (PO2 por debajo de 85 mmHg). Los pacientes con debilidad orofaríngea requieren intubación aún más temprana para prevenir la aspiración, pero en circunstancias más tardías la ventilación mecánica no siempre es requerida. Estos tratamientos demandan la admisión de los pacientes en una UTI donde puedan ser