BIOLOGIA Y GENETICA MOLECULAR 2.
2)Regulador de la conductancia transmembrana de la FQ. La CFTR es una proteína de 170.000 dalton anclada en la membrana por dos dominios transmembrana (TM-1 y TM-2) y cada dominio transmembrana atraviesa 6 veces a doble capa lipídica. Tiene dos sitios de unión al ATP (NBF1 y NBF2) y un dominio regulador (R) de alto contenido en aminoácidos eléctricamente cargados como glutámico, aspártico, glutamina y lisina (ver transparencia 2). TM-1 es el soporte físico del poro del canal. Este canal está regulado por estímulos hormonales, cuyo efecto se ejerce elevando la concentración intracelular de AMPc. El AMPc es un segundo mensajero que activa una proteína quinasa A (PK A), la cual a su vez fosforila a otras proteínas, que son activadas o inactivadas por esta fosforilación.
Existen 9 sitios “consenso” para fosforilación por PK A en el dominio R de la proteína CFTR. La apertura del canal CFTR se activa por fosforilación del dominio R por la PK A. Además es necesaria la hidrólisis de ATP por el NBF1 y que otra molécula de ATP se una al NBF2 para estabilizar la apertura del canal (ver transparencia 3).
Actualmente se sabe que el canal afectado en la FQ es CFTR (cuyo gen pertenece a la superfamilia de genes ABC, que en humanos incluye aquellos genes cuyos productos son proteínas de membrana cuya función es el transporte de sustancias a través de la membrana mediante un proceso dependiente de energía).
3)Mutaciones del gen.
Hasta la fecha se han identificado más de 620 mutaciones diferentes, responsables de la amplia manifestación fenotípica de la enfermedad. La mutación más frecuente es una deleción de tres pares de bases que determina la pérdida de la fenilalanina en posición 508 de la proteína codificada por CFTR. La frecuencia de esta mutación varía mucho según el grupo étnico y la localización geográfica. En la población española, la frecuencia de esta mutación representa el 53%.
La segunda mutación más frecuente es la G542X (frecuencia de un 8.3% en la población española).
Métodos de detección de mutaciones: son los mismos que se utilizan para otras patologías (directos o indirectos):
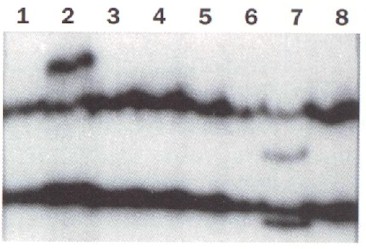
Polimorfismos de conformación de cadena simple (SSCA) y electroforesis en geles de gradiente denaturantes (DGGE): en ambos se obtiene un patrón de bandas anómalo si existe algún cambio en el fragmento de ADN que estamos estudiando.
En la figura de la página anterior observamos 8 muestras de individuos afectos (analizadas mediante SSCA). Los carriles 2 y 7 muestran dos fragmentos anómalos, que después de ser secuenciados correspondían a las mutaciones R117H y DE115.
*Correlación genotipo/fenotipo (ver transparencia 4): La expresión de la FQ es muy heterogenea. Dado el amplio espectro de mutaciones encontradas y las subsiguientes consecuencias moleculares debería existir correlación entre diferentes genotipos y sus fenotipos.
Se han realizado estudios agrupando pacientes que presentaban características clínicas comunes entre individuos con FQ con el mismo genotipo. De entre todos los parámetros analizados sólo la función pancreática se correlaciona bien con los fenotipos clínicos y parece correlacionar con diferentes mutaciones en CFTR. El factor limitante a la hora de establecer este tipo de correlaciones estriba en conseguir individuos homocigotos para una mutación determinada.
Una de las hipótesis que ha ido tomando más fuerza a la hora de explicar las diferencias encontradas entre individuos, que perteneciendo a la misma familia y presentando el mismo genotipo para CFTR poseen diferente fenotipo, es la existencia de un gen modulador de CFTR.
4)Detección de portadores.
Estudios moleculares en familias con FQ: Los trabajos con sondas moleculares estrechamente ligadas al gen, permitieron realizar los primeros estudios de portadores mediante el estudio de polimorfismos moleculares (FRLP: fragmentos de restricción de longitud polimórfica). Estos FRLPs permitían seguir la segregación del gen anómalo a lo largo de varias generaciones. A finales de los 80, la PCR simplificó mucho los estudios de segregación.
El screening se puede realizar en recién nacidos, jóvenes y mujeres embarazadas (se estudia a la embarazada, y sólo en caso aquellos casos positivos se pasa a estudiar a la pareja. En ciertas ocasiones se realizan estudios en cascada, pues una persona o pareja sin mutación conocida tiene todavía un riesgo residual elevado de tener un hijo con FQ.
La identificación de portadores es factible en la práctica totalidad de las familias con FQ (99%) combinando el estudio directo del gen con los estudios indirectos de sondas y microsatélites.